
Suscribirse


lunes, 19 de octubre de 2020
viernes, 2 de octubre de 2020
Vídeo del fin de semana
https://www.youtube.com/c/TodoomedicinaBlogspotScc
https://www.facebook.com/groups/2999973646724951/permalink/3479218795467098/?sfnsn=scwspwa
lunes, 21 de septiembre de 2020
Síndrome trombotico del COVID 19

domingo, 20 de septiembre de 2020
�� METRONIDAZOL ��
Vídeo donde platicamos un rato sobre el metronidazol, sobre su mecanismo de acción, que microorganismos ataca, cuales son sus principales aplicaciones clínicas y cuales los efector adversos característicos de este importante fármaco. Quieres saber más no te pierdas este vídeo.
👇👇👇 Más información de interés 👇👇👇
Les agradezco haber pasado por el canal.
Que tengan un día lleno de éxito.🙌
⭐ SUSCRIBETE ⭐ 👍 DALE LIKE 👍 ✒ COMENTA ✒
💎 Twitter: https://twitter.com/MI_Scc Sígueme también en Twitter y construyamos conocimiento juntos.
💣 https://www.facebook.com/groups/2999973646724951/ Grupo de Facebook sobre medicina interna, únete para que seas parte de esta hermosa comunidad.
📪 salvador.contreras.cornejo@gmail.com Necesitas escribirme algo en privado, hazlo por este medio.
💫 BLOGGER https://medicinainternascc.blogspot.com/ Conoce otros temas de interés como: 👉 CURSO COMPLETO DE MEDICINA TRANSFUSIONAL https://medicinainternascc.blogspot.com/2020/05/curso-de-medicina-transfusional.html
👉 CURSO COMPLETO DE ELECTROCARDIOGRAMA https://medicinainternascc.blogspot.com/2020/05/curso-de-electrocardiograma-basico.html
🔨INSTAGRAM https://www.instagram.com/medicina_interna_mx/
🔎WEB SITE https://dr-contreras.negocio.site/
lunes, 17 de agosto de 2020
NEUROPATÍA DIABÉTICA
NEUROPATIA
DIABETICA
Entre las complicaciones de la diabetes, un
grupo de síndromes clínicos causados por daños en los sistemas nerviosos
periférico y autónomo son, con mucho, los más prevalentes. Generalmente
conocidos como diferentes formas de neuropatía, estos síndromes son causados
por daño difuso y focal del sistema nervioso y ocurren hasta en la mitad de
todos los individuos con diabetes.
La forma más común de neuropatía diabética, la
polineuropatía simétrica distal.
La polineuropatía simétrica distal se
manifiesta con una distribución en "media y guante", por lo que las
manos y las extremidades inferiores suelen verse afectadas.
Otras neuropatías difusas secundarias a la
diabetes pueden ocurrir (Fig. 1) e
incluyen la constelación de neuropatías autónomas, como la neuropatía cardíaca
autónoma, la dismotilidad gastrointestinal y la cistopatía e impotencia
diabética.
Las neuropatías focales, aunque menos
frecuentes, incluyen la disfunción de los nervios periféricos individuales que
conducen a mononeuropatías aisladas o, con menor frecuencia, a las raíces
nerviosas que producen radiculopatía o polirradiculopatía.
Sin una intervención exitosa, se estima que de
los 9,7 mil millones de personas que se espera que vivan en 2050, un tercio
tendrá diabetes y la mitad de ellos tendrá neuropatía.
La neuropatía autonómica diabética engloba un
grupo de trastornos causados por el deterioro del sistema nervioso simpático
y parasimpático. La neuropatía autonómica cardíaca (CAN) puede presentarse como
debilidad generalizada, mareo o síncope franco acompañado de taquicardia o
bradicardia ortostática e intolerancia al ejercicio. Los síntomas de disfunción
autónoma gastrointestinal (también conocida como gastroparesia) incluyen
náuseas, distensión abdominal, saciedad temprana con poco apetito, vómitos
posprandiales y diabetes frágil (es decir, diabetes difícil de controlar). La
disfunción esofágica también puede ocurrir con disfagia (dificultad para
tragar) por alimentos sólidos y pirosis secundaria al reflujo ácido. La
neuropatía autonómica urogenital se presenta como una disfunción de la vejiga
(también conocida como cistopatía diabética) que puede variar desde retención
urinaria con vacilación hasta incontinencia urinaria con urgencia. La
disfunción sexual es otra manifestación común de la neuropatía autónoma
urogenital. En los hombres, la disfunción sexual se manifiesta como impotencia,
disminución de la libido y eyaculación anormal, mientras que en las mujeres, la
disfunción sexual se presenta como dolor durante el coito, falta de lubricación
y reducción de la libido. La disfunción suodomotora autónoma se presenta como
piel seca (anhidrosis) con sudoración gustativa. Mala lubricación y reducción
de la libido.
El tratamiento de la neuropatía autonómica
diabética depende del subtipo específico. Se recomienda optimizar el control de
la glucosa en las primeras etapas del curso de la diabetes mellitus tipo 1
(T1DM) para prevenir o retrasar la CAN, mientras que dirigirse a todos los
factores de riesgo metabólico es la recomendación para la diabetes mellitus
tipo 2 (T2DM). La repleción de volumen, la actividad física, la fludrocortisona
o midodrina en dosis bajas y las medias de compresión se encuentran entre las
opciones de tratamiento para la CAN en pacientes con DM1 o DM2.
El tratamiento farmacológico de la disfunción
eréctil masculina incluye inhibidores de la fosfodiesterasa tipo 5. El fármaco
antimuscarínico tópico glicopirrolato puede usarse para el tratamiento de la
sudoración gustativa, mientras que las lociones humectantes diarias brindan
alivio a la piel seca. Una revisión exhaustiva de la neuropatía autonómica
diabética229 junto con pautas de tratamiento detalladas 9 pueden proporcionar
al lector una discusión más profunda del tema.
Epidemiología
Aumentar las caídas, provocar dolor y reducir
la calidad de vida (CV) 9 . Los costos anuales de la neuropatía diabética y sus
complicaciones superan los $ 10 mil millones en los Estados Unidos.
Prevalencia de 1% a 4% para la neuropatía, con
40% a 55% de estos casos secundarios a diabetes.
Países Bajos, la incidencia de neuropatía
aumenta drásticamente con los años, de <50 casos por 100.000 personas-año en
los <50 años a ~ 300 por 100.000 personas-año en los> 75 años, y la
diabetes representa la 32% de todos los casos.
La incidencia de neuropatía es mayor en
personas con DMT2 (6.100 por 100.000 personas-año) que en aquellas con DMT1
(2.800 por 100.000 personas-año).
La prevalencia de neuropatía es similar en
aquellos con DMT2 (8-51% a aquellos con DMT1 (11-50%.
Factores de riesgo
La duración de la diabetes y los niveles de
hemoglobina A 1c son los principales predictores.
Independiente de hba 1cniveles, el número de
componentes del síndrome metabólico, como hipertrigliceridemia, hipertensión,
obesidad abdominal y niveles bajos de lipoproteínas de alta densidad (HDL), se
asocia constantemente con la neuropatía diabética en pacientes con DM2.
Otros factores de riesgo independientes para
el desarrollo de la neuropatía diabética incluyen el tabaquismo, el abuso de
alcohol, el aumento de estatura y la edad avanzada.
Varios genes están relacionados con la
neuropatía diabética, pero solo los polimorfismos ACE (que codifica la enzima
convertidora de angiotensina) y MTHFR (que codifica la metilentetrahidrofolato
reductasa) se han estudiado en múltiples poblaciones, incluidas grandes
cohortes.
Mecanismos / fisiopatología
La neuropatía diabética es un trastorno
neurodegenerativo único del sistema nervioso periférico que se dirige
preferentemente a axones sensoriales, axones autónomos y más tarde, en menor
medida, axones motores.
La neuropatía diabética progresiva implica la
retracción y la "muerte" de los axones sensoriales terminales en la
periferia, con relativa preservación de los perikarya (cuerpos celulares).
"media y guante" refleja primero el
daño de los axones sensoriales más largos, por ejemplo, la pérdida de los
axones epidérmicos distales de la pierna antes de la pérdida de las
extremidades más proximales; por esta razón, la neuropatía diabética se
considera una neuropatía dependiente de la longitud.
El orden exacto de la lesión celular (si, por
ejemplo, Actualmente se desconoce el daño a las células de Schwann o axones
antes de que se produzcan daños en los cuerpos de las células neuronales) en la
diabetes. Estos cambios incluyen alteraciones en el transporte de
células-axones de Schwann, alteraciones en la expresión de proteínas en los
GRD, desmielinización y degeneración. GAP43, proteína 43 asociada al
crecimiento; HSP, proteína de choque térmico; PARP, poli (ADP-ribosa) polimerasa.
Aunque la neuropatía diabética no se considera
principalmente una neuropatía desmielinizante, las células de Schwann son el
blanco de la hiperglucemia crónica, y los casos más graves de neuropatía
diabética en pacientes incluyen características de desmielinización.
Dado el estrecho e íntimo apoyo mutuo entre
los axones y las células de Schwann, el daño de las células de Schwann podría
conducir a varias alteraciones en el axón.
Se ha propuesto que la expresión reducida de
arnm que codifica el neurofilamento subyace a esta pérdida de polímeros de
neurofilamento.
También asocian el estrés del retículo
endoplásmico con el daño del nervio periférico mediado por la diabetes 42eso
afectaría la función nerviosa.
La hiperglucemia altera la función de
moléculas de plasticidad clave, como la proteína 43 asociada al crecimiento
(GAP43; también conocida como neuromodulina) y la β-tubulina, y los patrones de
expresión del choque térmico. Proteínas (HSP) 43 , 44 y poli (ADP-ribosa)
polimerasa (PARP) 45 , 46 en el DRG.
Los datos sugieren que la disfunción en estas
vías promueve el procesamiento anormal de proteínas, el daño oxidativo y la
disfunción mitocondrial, lo que lleva a la pérdida de la función nerviosa
periférica.
Se ha informado sobre la regulación al alza de
las vías implicadas en la inflamación, la bioenergética y el procesamiento de
lípidos en matrices de nervios ciáticos de modelos preclínicos de T1DM y T2DM.
Hiperglucemia e hiperlipidemia.
En las
células de Schwann, las neuronas DRG y los axones, tanto la glucosa como los
ácidos grasos producen NADH y FADH 2 a través de la glucólisis y el ciclo del
ácido tricarboxílico (glucosa) y la β-oxidación (ácidos grasos). Cuando los
ácidos grasos de cadena larga se transportan a las células de Schwann para
someterse a una β-oxidación, cada ciclo de β-oxidación forma una molécula de
acetil-coa, que se transporta al ciclo del ácido tricarboxílico para NADH y
FADH 2formación. Sin embargo, durante la sobrecarga de sustrato, como en la
diabetes, el sistema de transporte se satura y las moléculas de acetil-coa se
convierten en acilcarnitinas. La acumulación de acilcarnitinas es tóxica tanto
para las células de Schwann como para las neuronas DRG, lo que se suma a la
lesión en curso del sistema nervioso en la neuropatía diabética. Las
acilcarnitinas acumuladas se liberan de las células de Schwann y pueden inducir
la degeneración axonal, que se ha propuesto que implica una disfunción
mitocondrial y una respuesta de estrés integrada desadaptativa en las células
de Schwann.
NADH y FADH 2 se transportan en las
mitocondrias a través de los complejos I-IV para producir ATP a través de la
fosforilación oxidativa. Un subproducto de la fosforilación oxidativa es la
producción de niveles bajos de especies reactivas de oxígeno (ROS) que son
fácilmente neutralizadas por antioxidantes celulares innatos, como superóxido
dismutasa, glutatión y catalasa 58 , 59 , 60 , 61 , 62 , 63 . Sin embargo,
durante el exceso de carga de sustrato, como en la diabetes, falla la
fosforilación oxidativa, lo que conduce a la pérdida de producción de ATP y al
aumento de los niveles de ROS, lo que posteriormente conduce a insuficiencia
mitocondrial y daño metabólico y oxidativo de las células de Schwann y las
neuronas DRG-
Las
mitocondrias disfuncionales producen energía insuficiente y pierden la
capacidad de conducir normalmente hacia abajo de los axones, lo que promueve
aún más la alteración y lesión axonal.
El aumento de los niveles de glucosa conduce
al metabolismo de la glucosa a través de las vías de los poliol y la
hexosamina, lo que da como resultado un aumento de ROS e inflamación,
respectivamente, en gran parte debido a la lesión mitocondrial 37 , que
contribuye a la disfunción continua del sistema nervioso.
El aumento de los niveles de glucosa conduce a
la glicación de numerosas proteínas estructurales y funcionales para producir
productos finales de glicación avanzada (AGE). Los AGE dan como resultado una
alteración o pérdida de la función proteica e interactúan con el receptor
específico de AGE (RAGE) para modificar la expresión génica y la señalización
intracelular y promover la liberación de moléculas proinflamatorias y radicales
libres.
El exceso de ácidos grasos libres catabolizados
por la β-oxidación en respuesta a la hiperlipidemia puede dañar el sistema
nervioso periférico, particularmente las células de Schwann.69 , a través de la
generación de ROS e inflamación sistémica y local a través de la activación de
macrófagos con posterior producción de citocinas y quimiocinas.
La oxidación de colesterol a Oxisteroles en la
lesión del tejido neuronas actúa de mediador 58 , 71 , mientras que las
lipoproteínas del plasma, en particular de lipoproteínas de baja densidad
(LDL), son oxidados por ROS y se unen receptor de LDL oxidada 1 (LOX1) (ref. 72
), Toll-like receptor 4 (TLR4) 73 y RAGE 74 . La unión de las LDL oxidadas a
estos receptores activa una serie de cascadas de señalización, incluida la
activación de la caspasa 3 y la degradación del ADN nuclear 74 , que median la
inflamación adicional y la acumulación de ROS, con lesión nerviosa continua y
progresiva.
Contribuciones microvasculares
Las deficiencias en el suministro de sangre a
los nervios periféricos se consideran un posible mecanismo patológico adicional
de la neuropatía diabética-
La disfunción microcirculatoria está
fuertemente asociada con la disfunción del nervio periférico y se ha propuesto
un ciclo de microcirculación deficiente que conduce a daño adicional del
nervio. Los aumentos en la densidad capilar endoneural están presentes en
pacientes con diabetes en comparación con individuos sanos, lo que sugiere que
la densidad capilar puede responder a la isquemia nerviosa inducida por la
diabetes.
Se ha informado de una vasodilatación
deficiente de las arteriolas epineuriales en ratas diabéticas, y este cambio
aparece antes de la disminución de la NCV.
La insulina administrada cerca del nervio, o
en la piel plantar donde accede a los axones dérmicos, también repara las anomalías
de la diabetes en modelos animales experimentales.
Mecanismos del dolor
El dolor neuropático se define como el dolor
causado por una lesión o enfermedad del sistema nervioso somatosensorial.
Aproximadamente el 30-50% de los pacientes con neuropatía diabética desarrollan
dolor neuropático-
La alodinia provocada por el cepillado (cuando
un estímulo normalmente no nocivo evoca dolor) y parestesias.
Factores de riesgo de la neuropatía diabética
dolorosa
El sexo femenino es un factor de riesgo de
neuropatía diabética dolorosa-
Control glucémico deficiente 96 , la función
renal alterada 95 y el índice de masa corporal (IMC) alto.
Hiperexcitabilidad de las neuronas sensoriales
Las neuronas sensoriales lesionadas, como en
la neuropatía diabética, desarrollan hiperexcitabilidad y pueden generar
potenciales de acción en ausencia de un estímulo (actividad espontánea) y
desarrollar una función estímulo-respuesta alterada 97 , 98 , 99 (Fig. 4 ). Esta actividad aberrante es crucial para
el mantenimiento del dolor neuropático, incluso en pacientes con dolor
prolongado.
La neuropatía diabética dolorosa se asocia con
importantes cambios psicológicos y comorbilidades, que incluyen un aumento de
la ansiedad, depresión y trastornos del sueño.
Diagnóstico, cribado y prevención
Diagnóstico
La neuropatía diabética es la presencia de
síntomas y / o signos de disfunción del nervio periférico en pacientes con
diabetes después de descartar otras etiologías.
Para la gran mayoría de los pacientes, el
diagnóstico de neuropatía diabética se basa únicamente en la historia y el
examen y no se necesitan pruebas adicionales-
Los síntomas de la neuropatía diabética son
entumecimiento, hormigueo, dolor y debilidad e inestabilidad, que comienzan
distalmente (en los dedos de los pies) y se extienden proximalmente y luego a
los dedos de las extremidades superiores cuando los síntomas de las
extremidades inferiores llegan a las rodillas.
Los pacientes a menudo tienen una neuropatía
predominantemente de fibras pequeñas en las primeras etapas del curso de la
neuropatía diabética o cuando se les diagnostica prediabetes 134 , y tienen
síntomas dolorosos distales de dolor quemante, lancinante y congelante que son
mayores en reposo. La lesión por fibras grandes suele ocurrir más tarde en el
curso de la enfermedad, pero no siempre es así.
Los hallazgos clínicos de la neuropatía
diabética son la pérdida de la sensibilidad al pinchazo, la temperatura
(principalmente frío), la vibración y la propiocepción en una distribución de
"calcetines y guantes".
Las sensaciones de pinchazo y temperatura
están mediadas por pequeñas fibras nerviosas, mientras que la sensación de
vibración y la propiocepción están mediadas por grandes fibras nerviosas.
La pérdida de los reflejos del tobillo se
produce al principio de la neuropatía diabética; por tanto, el examen inicial
debe incluir pruebas de reflejos. Posteriormente, se observa debilidad de los
músculos pequeños del pie y dorsiflexores.
Los síntomas y signos clínicos de la
neuropatía diabética se pueden combinar en escalas, como en el Toronto Clinical
Neurothy Score 135 , el Toronto Clinical Neurothy Score modificado 136 o el
Michigan Diabetic Neurothy Score 137 , que tienen valores de corte definidos
para la presencia de neuropatía.
Los cambios en la NCS en pacientes con
neuropatía diabética incluyen amplitudes disminuidas, velocidades de conducción
disminuidas y respuestas F prolongadas
Los NCS son normales en pacientes con
neuropatía principalmente de fibras pequeñas, y estos pacientes típicamente
también tienen un examen clínico casi normal 140 . El estándar de oro para el
diagnóstico de la neuropatía de fibras pequeñas es la medición de la densidad
de fibras nerviosas intraepidérmicas (IENFD) por biopsia cutánea con punch.
Las declaraciones de posición actuales de la
Asociación Americana de Diabetes (ADA) y las pautas de la Asociación Canadiense
de Diabetes recomiendan la detección de neuropatía diabética en el momento del
diagnóstico y anualmente para pacientes con DM2 y 5 años después del
diagnóstico y luego anualmente para pacientes con DMT1
La prueba de monofilamento de 10 g se puede
utilizar para predecir la neuropatía diabética 154 incidente . El valor de este
monofilamento es que las insensibilidades más altas predicen un alto riesgo de
ulceración del pie
La prueba de vibración con un diapasón de 128
Hz (cronometrada o la cantidad de veces que se siente) tiene capacidades de
discriminación similares a la prueba de monofilamento y también es rápida y
fácil de realizar
La evaluación de los reflejos tendinosos profundos
tiene buenas características de prueba, aunque no tan altas como las pruebas de
monofilamento o vibración.
El tratamiento de la hiperglucemia sería
lógicamente el mejor tratamiento preventivo de la neuropatía diabética.
Potencial del ejercicio para prevenir lesiones
nerviosas e incluso promover la regeneración nerviosa,
DCCT / EDIC, el control intensivo de la
glucosa retrasó significativamente su desarrollo y progresión a lo largo del
tiempo
El ensayo ALADIN III multicéntrico,
aleatorizado, doble ciego y controlado con placebo demostró una mejora
significativa en la puntuación de deterioro de la neuropatía (NIS) en pacientes
que recibieron ácido α-lipoico pero sin una mejora significativa en la
puntuación total de síntomas (TSS)
Ensayo SYDNEY2, 181 pacientes con neuropatía
diabética recibieron dosis orales una vez al día de 600 mg, 1200 mg o 1800 mg
de ácido α-lipoico o placebo durante 5 semanas 177 . La medida de resultado
primaria fue el cambio desde el inicio del TSS, que disminuyó en un 51% en el
grupo de 600 mg, un 48% en el grupo de 1200 mg y un 52% en el grupo de 1800 mg
en comparación con el 32% en el grupo de placebo
Epalrestat se comercializa en Japón para el
tratamiento de la neuropatía diabética. Es un inhibidor de la aldosa
reductaasa.
Los inhibidores de la recaptación de
serotonina y noradrenalina (IRSN) y los antidepresivos tricíclicos (ATC) tienen
la mejor evidencia para respaldar su uso en el tratamiento del dolor
neuropático diabético.
Anticonvulsivos
De los anticonvulsivos, los ligandos a2 δ
gabapentina y pregabalina son eficaces para la neuropatía diabética dolorosa.
La mayoría de los estudios de pregabalina
muestran eficacia en la neuropatía diabética dolorosa, con al menos un 30-50%
de mejoría del dolor
Los efectos adversos de ambos medicamentos
pueden incluir confusión y mareos, son más graves en los pacientes mayores 197
y pueden atenuarse con dosis iniciales más bajas y una titulación más gradual.
SNRI
La duloxetina es un IRSN selectivo con
eficacia demostrada para el tratamiento de la neuropatía diabética dolorosa en
varios ensayos aleatorizados multicéntricos 9 , 198 , 199 . El tratamiento con
duloxetina también puede mejorar la calidad de vida relacionada con la neuropatía
200 , 201 . La venlafaxina es otro IRSN que puede ser eficaz en el tratamiento
del dolor en la neuropatía diabética 202 , 203 . Los IRSN se asocian con una
serie de efectos adversos que pueden ser más graves que los observados con
gabapentina y pregabalina, como mareos, fatiga, náuseas e insomnio.
TCA: La amitriptilina es el ATC más utilizado
y ha demostrado eficacia en la neuropatía diabética dolorosa en pequeños
ensayos clínicos aleatorizados, ciegos y controlados con placebo 204 , 205 .
Nortriptilina y desipramina tienen menos efectos adversos que la amitriptilina
y la imipramina y podrían ser más seguras en adultos mayores
Analgésicos opioides y opioides atípicos
Aunque existe evidencia de la eficacia de los
opioides para aliviar el dolor, estos medicamentos están asociados con un alto
riesgo de adicción y problemas de seguridad; por lo tanto, la declaración de
posición más reciente de la ADA no recomienda el uso de opioides como terapias
de primera o segunda línea para tratar el dolor neuropático asociado con la
neuropatía diabética
Dos grandes estudios han demostrado la
eficacia del tramadol para la neuropatía diabética dolorosa 214 , 215 , y el
efecto podría ser duradero
Hay varias herramientas psicométricas
disponibles para evaluar el efecto de la diabetes y sus complicaciones en la
vida de los pacientes, así como el efecto de las intervenciones médicas. Estos
incluyen la medida Diabetes Quality of Life (DQOL), la Diabetes-Specific
Quality of Life Scale (DSQOLS), la Escala de evaluación de la diabetes, el
ATT-39, el Cuestionario sobre el estrés en pacientes con diabetes revisado, la
Diabetes tipo 2 Lista de verificación de síntomas, Escala de áreas
problemáticas en la diabetes (PAID-1) y Auditoría de la calidad de vida
dependiente de la diabetes (addqol).
Una nueva forma de mejorar la calidad de vida
en personas con neuropatía diabética dolorosa es utilizar la terapia
cognitivo-conductual (TCC). La TCC puede ayudar a reducir la intensidad del
dolor y mejorar la función física. De hecho, la TCC tuvo un efecto beneficioso
sobre el dolor crónico mixto y la calidad de vida en un estudio reciente 228 .
Diez sesiones semanales de TCC grupal de 90 minutos que incluían refuerzo
motivacional y entrenamiento destinado a reducir la intensidad del dolor y la
depresión tuvieron un beneficio duradero positivo en la calidad de vida del
paciente durante un máximo de 6 meses.
VÍDEO RECOMENDADO:
jueves, 13 de agosto de 2020
ENFERMEDAD RENAL DIABETICA "NEFROPATÍA DIABETICA"
INTRODUCCION
La nefropatía diabética es la principal causa de ERC
terminal en los EUA.
La DT representa 30 al 50% de los casos de ERC de nuevo
diagnostico en EUA.
Solo 30 a 40 % de los pacientes con DT desarrollan
nefropatía diabética.
El tratamiento se divide en 4 áreas:
1.-
Reducción del riesgo cardiovascular.
2.-
Control Glucémico.
3.-
Control de la Presión Arterial.
4.-
Inhibición del SRAA.
Metas:
1.- HbA1c
menor a 7 %
2.- TA
< 140/90 mmHg.
Epidemiología.
La
nefropatía diabética es un diagnóstico que se refiere a cambios patológicos
específicos estructurales y funcionales observados en pacientes con DM, cambios
que se traducen en proteinuria, hipertensión y reducciones progresivas de la
función renal.
El
riesgo de desarrollar ND tiene un componente genético, poli génico.
Los
afroamericanos, nativos americanos y mexicoamericanos tienen un riesgo mayor
respecto a americanos europeos.
Hay
agrupación familiar. Tener padres con nefropatía diabética te confiere un mayor
riesgo.
Genes
candidatos: Transportador 2 de glucosa, factor de crecimiento transformante
beta, sintasa de óxido nítrico endotelial.
Fisiopatología.
Es resultante de la generación y circulación de productos finales de la glicación avanzada (Los productos finales de glicación (AGEs) son un grupo heterogéneo de moléculas generadas por medio de reacciones no enzimáticas de glicación y de oxidación de proteínas, lípidos y ácidos nucleicos (reacción de Maillard), elaboración de factores de crecimiento, cambios hemodinámicos y hormonales. Estos conducen a la liberación de especias reactivas de oxígeno y mediadores inflamatorios, En conjunto estos cambios resultan en hiperfiltración glomerular, hipertensión glomerular, hipertrofia renal y composición glomerular alterada que se manifiesta como albuminuria e hipertensión.









Desde el punto de vista patológico, los riñones sufren
varios cambios, que incluyen el depósito (principalmente en el mesangio) de
matriz extracelular, engrosamiento de la membrana basal glomerular, cambios
proliferativos y atrofia tubular, que en última instancia dan como resultado
fibrosis intersticial y glomeruloesclerosis.
Con la aparición de la DM, el tamaño y el peso de los
riñones aumentan en promedio un 15%, y este aumento de tamaño permanece incluso
después de que se produzcan reducciones progresivas en la función renal.
La lesión patológica clásica de la nefropatía diabética es
de naturaleza nodular y fue descrita por primera vez por Kimmelstiel y Wilson
en 1936. Los nódulos son típicamente acelulares y positivos por la tinción de
ácido periódico-Schiff. Aunque estos nódulos son patognomónicos para la
nefropatía diabética, se notifican en sólo el 10% al 50% de las muestras de
biopsia de pacientes con T1 / T2DM.
Mucho más común es la lesión glomerular difusa que se
caracteriza por expansión difusa de la matriz mesangial. Las lesiones
arteriolares que afectan tanto a los vasos aferentes como a los eferentes
también son prominentes y frecuentes en la DM.
Kussman et al: la proteinuria aparece de 11 a 23 años
después del diagnóstico de T1DM, la concentración de creatinina sérica comienza
a aumentar después de 13 a 25 años y la enfermedad renal en etapa terminal se
desarrolla después de 18 a 30 años. Con el posterior desarrollo de ensayos más
sensibles para detectar la albúmina urinariaexcreción, se observó que pequeñas cantidades de
albúmina en la orina (microalbuminuria; 30-300 mg / g de creatinina) preceden
al desarrollo de proteinuria manifiesta (macroalbuminuria;> 300 mg / g de
creatinina) en la mayoría de los pacientes, que ocurre de 5 a 10 años después
de la diagnóstico de DM.
El mayor predictor de deterioro de la función renal y
progresión de la nefropatía diabética es la proteinuria.
Cuando ha comenzado la pérdida de la función renal, como lo
demuestra un aumento de la concentración de creatinina sérica o una disminución
de la tasa de filtración glomerular estimada (eGFR), el paciente con nefropatía
diabética comienza un descenso continuo hacia la insuficiencia renal crónica y
la terapia de reemplazo renal o la muerte.
The mean duration of life after the onset of proteinuria was 4.8 years, the longest survival being 13.3 years. The mean survival after the onset of early renal failure was 2.7 years, with the longest survival being 5.7 years. The time between the onset of late renal failure to death was only seven months, and no patient survived more than 12 months. The mortality for the entire group was 53%. Renal failure was the cause of death in 59% of fatal cases, and cardiovascular disease was responsible for 36%.
Según estudios de pacientes no tratados con DMT1 y de indios pima con DMT2, la tasa de pérdida de la TFG puede ser del orden de 7 a 12 ml / min / 1,73 m 2 por año. El tratamiento con inhibidores del sistema renina-angiotensina (RAS) ha reducido esta tasa de disminución a 3 a 6 ml / min / 1,73 m 2por año.
Diagnóstico de nefropatía diabética.
El desarrollo de albuminuria significativa antes de 5 años o
después de 25 años de duración de la DM1 disminuye la probabilidad de
nefropatía diabética. Además, el 95% de los pacientes con DM1 y nefropatía
diabética también tienen retinopatía diabética., por lo que la ausencia de
retinopatía puede implicar un diagnóstico diferente al de nefropatía diabética.
los pacientes con DM2son más desafiantes porque estas pistas epidemiológicas no son tan útiles. La retinopatía diabética es concordante con la nefropatía diabética en solo alrededor del 60% al 65% de los casos; por lo tanto, su ausencia no genera un alto valor predictivo negativo para el diagnóstico de nefropatía diabética.
La DKD se identifica clínicamente por una relación de
albúmina / creatinina urinaria alta persistentemente ≥30 mg/g o una reducción
sostenida de la TFGe por debajo de 60 ml / min por 1,73 m 2.
El cribado para DKD debe realizarse anualmente para
pacientes con DM1 comenzando 5 años después del diagnóstico y anualmente para
todos los pacientes con DM2 comenzando en el momento del diagnóstico.
Tratamiento.
Diabetes Control and Complications Trial (DCCT). Este ensayo seminal, realizado de 1983 a 1993 en los Estados Unidos y Canadá, asignó aleatoriamente a 1.441 pacientes a un control glucémico intensivo (objetivo HbA 1c < 6,05%) versus convencional con un seguimiento de una media de 6,5 años. La concentración media de HbA 1c fue del 9,1% frente al 7,3% para el control convencional frente al intensivo. El control intensivo resultó en unareducción del riesgo relativo del 39% para el desarrollo de microalbuminuria y reducción del riesgo relativo del 56% para la proteinuria manifiesta.
Datos observacionales de 2 ensayos clínicos aleatorizados
que prueban una intervención en pacientes con DM2, IDNT (Irbesartan Diabetic Nephropathy
Trial) y RENAAL (Reducción de los criterios de valoración en la diabetes
mellitus no insulinodependiente con el antagonista de angiotensina II Losartan)
son conocidos por demostrar el beneficio de los bloqueadores de los receptores
de angiotensina (ARA) para retrasar la progresión de la enfermedad renal.
El bloqueo de RAS se ha estudiado en pacientes con T1 / T2DM
sin microalbuminuria para evaluar si la terapia puede prevenir su desarrollo.
Múltiples ensayos en pacientes con T1DM (RASS [Estudio del sistema de
renina-angiotensina], DIRECT [ Ensayo de candesartán de retinopatía diabética ]
-Prevent 1 y DIRECT-Protect 1) no demostraron un beneficio de la terapia para
prevenir el desarrollo de microalbuminuria. Estos resultados sugieren que la
terapia temprana en pacientes con T1DM es ineficaz para prevenir el desarrollo
de microalbuminuria.
Esta estrategia de tratamiento también se ha probado en
pacientes con DM2 con resultados mixtos. El uso de ramipril en el ensayo HOPE
(Heart Outcomes Prevention Evaluation) no fue eficaz para este propósito.
BENEDICT (el Bergamo Nephrologic Diabetes Complications Trial) asignó al azar a
los pacientes a 1 de 4 brazos (placebo, trandolapril, verapamilo o trandolapril
más verapamilo) durante al menos 3 años con un objetivo de PA < 120/80 mmHg. Los 2 brazos que contenían
trandolapril mostraron un beneficio en la prevención del desarrollo de
albuminuria, y los análisis post hoc sugirieron que el efecto era independiente
de la reducción de la PA. Por último, el ROADMAP ( olmesartán aleatorio and
Diabetes Microalbuminuria Prevention) siguió a 4.449 participantes durante una
mediana de 3,2 años. Hubo una diferencia de seguimiento estadísticamente
significativa en la PA entre los brazos de olmesartán y placebo. El análisis primario
del ensayo mostró que olmesartán previno o retrasó la aparición de
microalbuminuria, con microalbuminuria desarrollándose en 8.2% versus 9.8% de
los participantes.
Si algún bloqueo de RAS es bueno, como se señaló
anteriormente, ¿es mejor? La cuestión de la terapia con múltiples agentes que
bloquean el RAS se abordó en 3 grandes ensayos clínicos. El primero fue
ONTARGET ( Telmisartan en curso solo y en combinación con Ramipril Global
Endpoint), un ensayo de resultados cardiovasculares que asignó al azar a 25.620
pacientes con riesgo de enfermedad cardiovascular a ramipril, telmisartán o
ambos. No hubo diferencia entre los 3 brazos en el resultado cardiovascular
compuesto.
El estudio VA NEPHRON-D (Veterans Affairs Nephropathy in
Diabetes) asignó al azar a 1.448 participantes con DM2 y proteinuria manifiesta
a losartán, 100 mg, diariamente en
combinación con lisinopril , 40 mg,
diariamente o losartán, 100 mg, diariamente más placebo. Este ensayo finalizó
antes de tiempo debido a un aumento de los eventos adversos (lesión renal aguda
e hiperpotasemia) en el grupo de tratamiento combinado. ALTITUDE (Aliskiren
Trial in T2DM Using Cardio-Renal Endpoints) evaluó si el bloqueo dual de RAS
con aliskiren y un inhibidor de la ECA o un ARB reducía los eventos
cardiovasculares y renales. Este ensayo también se terminó antes de tiempo
debido a un aumento en los eventos adversos y ningún beneficio aparente en el
grupo de terapia dual.
martes, 4 de agosto de 2020
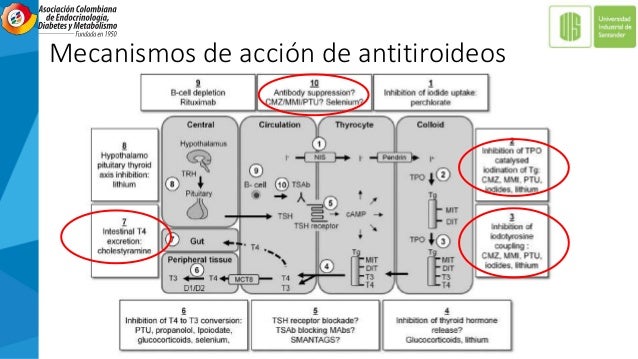
ENFERMEDAD DE GRAVES










-
 Se trata de un curso online basado en una recopilación de vídeos de youtube donde se habla desde las bases físicas del electrocardiograma co...
Se trata de un curso online basado en una recopilación de vídeos de youtube donde se habla desde las bases físicas del electrocardiograma co... -
Se trata de un pequeño curso en vídeo de youtube donde recopilo los vídeos más interesantes sobre el tema, si encuentras uno sugieremelo par...
-
NEUROPATIA DIABETICA Entre las complicaciones de la diabetes, un grupo de síndromes clínicos causados por daños en los sistemas nervio...

